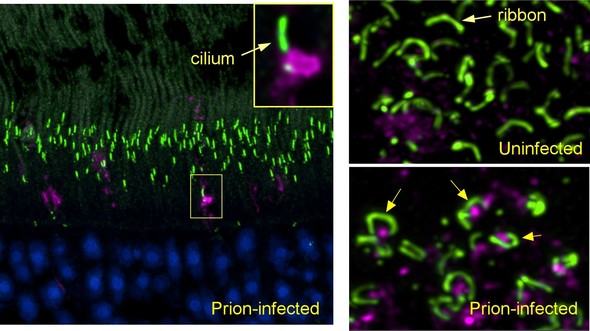
| Friday, January 29, 2021 (left panel) Early in prion infection, a prion protein aggregate (magenta) blocks the entrance to a cilium (green) in a retinal photoreceptor. (lower right) In prion-infected retina, prion protein (magenta) accumulates under the horseshoe-shaped ribbon synapses (green) found in photoreceptor terminals. (NIAID image) NIAID Scientists Identify Mechanism of Early Prion Disease in the Eyes The earliest eye damage from prion disease takes place in the cone photoreceptor cells, specifically in the cilia and the ribbon synapses, according to new NIAID research published in Acta Neuropathologica Communications. Prion diseases originate when normally harmless prion protein molecules become abnormal and gather in clusters and filaments in the human body and brain. Understanding how prion diseases develop, particularly in the eye because of its diagnostic accessibility to clinicians, can help scientists identify ways to slow the spread of these diseases. The scientists say their findings also may help inform research on human retinitis pigmentosa, an inherited disease with similar photoreceptor degeneration leading to blindness.  | 
No comments:
Post a Comment